 |
 |
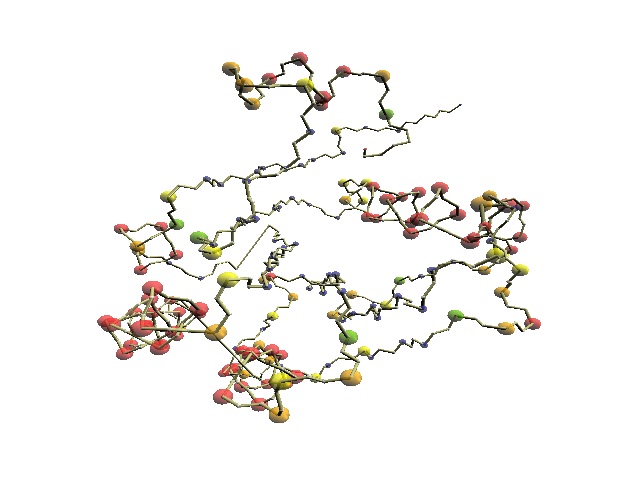
"Analysis of spacial structures of KH domains in hnRNP K and S3 reveals that they are topologically dissimilar and thus belong to different protein folds. ... The two distinct topologies might have arisen from an ancestral KH-motif protein by N- and C-terminal extensions, or one of the existing topologies may have evolved from the other by extension, displacement, and deletion." (Grishin, 2001) | |
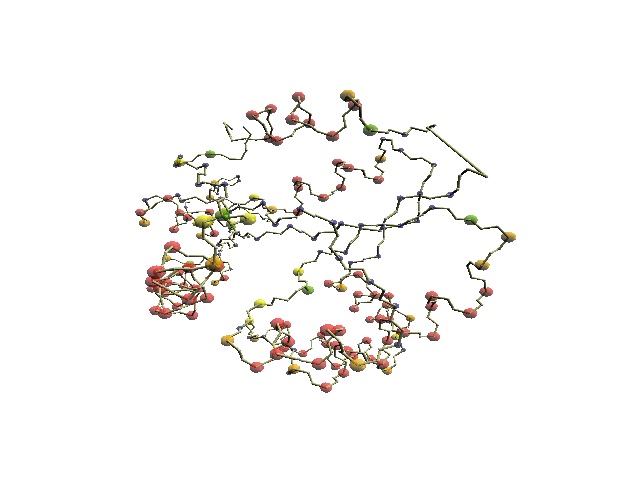
| 1j5e (ResC62-107) | 1khm
(ResA11-72) |
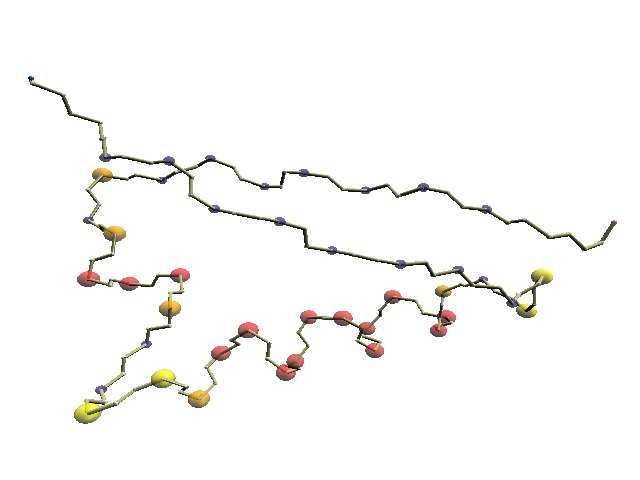
| 1j5e
VAVTVHVAKPGVV IGR GGERIRVLREELAKL
T
G K NVALNVQE code 0000000Q0QAAA B00 RRQAAAAAAAAAAAB 0 R R 00000000 LCS 0000000 0QAAA B0 R QAAAAAAAA B 0 R R 00000000 code 0000000 0QAAAAAAB0QBR QAAAAAAAA BR0000000000RGRG00000000QAAA 1khm ITTQVTI PKDLARSIIGKGG QRIKQIRHE SGASIKIDEPLEGSEDRIITITGTQDQI *RATIO : lcs/trimmed_seq_a = 0.857 (= 36/ 42), lcs/trimmed_seq_b = 0.632 (= 36/ 57) |
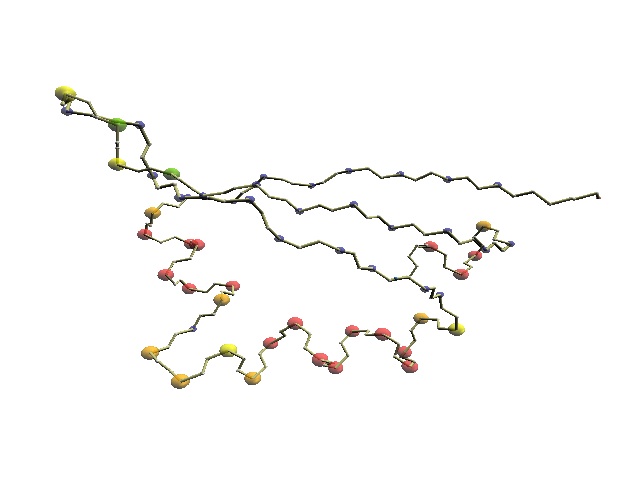
| Structural alignment by ComSubstruct |
| DSSP .LLLL.leeeelLHHHHHLLLLLHHHHHHHHHHlllllllLLEEEELL................... 1j5e .DNVA.vtvhvaKPGVVIGRGGERIRVLREELAkltgknvALNVQEVQ................... || || || | | | 1khm pIITTqvtipkdLARSIIGKGGQRIKQIRHESG......aSIKIDEPLegsedriititgtqdqiqn DSSP lLEEEeeeeehhHHHHHHLLLLHHHHHHHHHHL......lEEEELLLLlllleeeeeeeelhhhhhl *Z-score 1.1 AR 33 RMSD 3.3A SI 27% |
| Structural alignment by DaliLite |