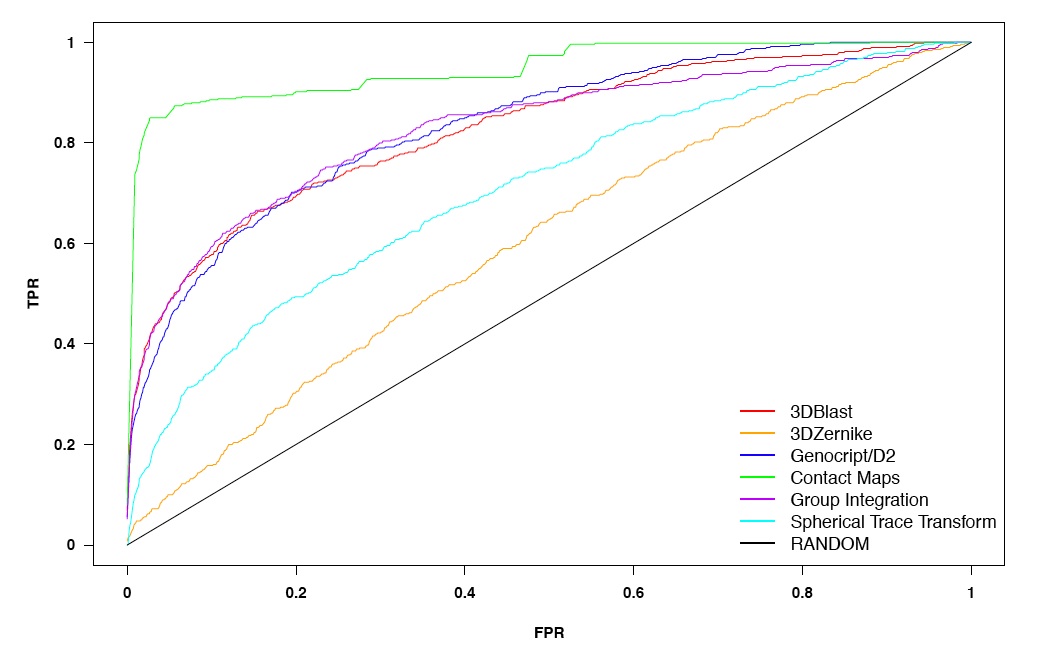
This table summarizes the
retrieval rates for all the methods. There were five cases in which
none of the methods found the nearest neighbour. These were: Q12
(1wwjA00), Q30 (1iicA02), Q40 (3c4aA01), Q43 (1nyaA00), and Q48
(3bioA02). In a further seven cases, only one method found the nearest
method as the top match.
However, there were 11 additional cases in which several methods found
the nearest neighbour as the second hit (i.e. 4 for GENOCRIPT, 3 for
Group Integration, 3 for 3DBlast and 1 for 3DZernike). |