"We show that structural
diversity has a significant effect on
structural alignment. Moreover, we observe alignment inconsistencies
even for modest spatial divergence, implying that the biological
interpretation of alignments is less straightforward than commonly
assumed. A salient example is the GroES 'mobile loop' where
sub-Ångstrom variations give rise to contradictory sequence
alignments." [1]
The D2 encoding method (, that is, programs ProteinEncoder and ComSubStruct, ) is compared with existing methods.
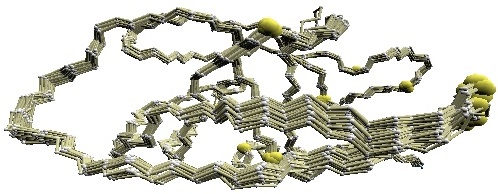
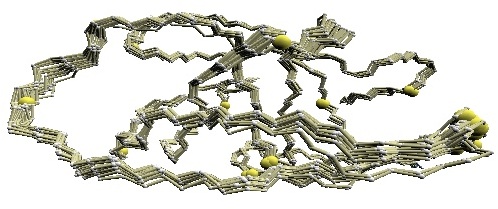
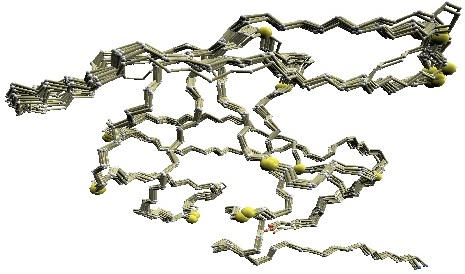
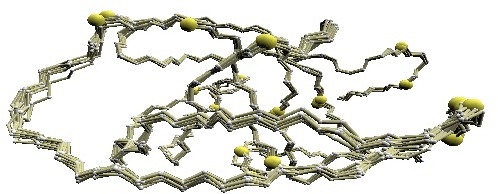
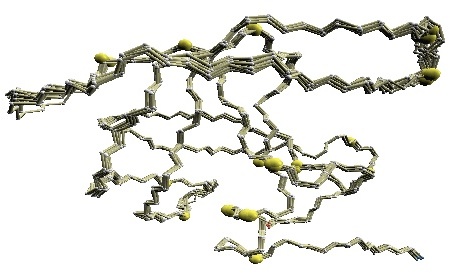
We used twelve X-ray crystal structures of GroES used in [1] for the comparative study: 1p3h (chain N), 1aon (chain O, P, and S), 1pcq (chain O, P, Q, R, S), 1pf9 (chain O and R), and 1svt (chain O).
(1.1) 1D representaion of protein backbone
The D2 code (ProteinEncoder) is compared with two encoding methods: HMM-SA (structural alphabet) [2] and Stride (secondary structure assignment) [3].
(1.2) Structural alignment
Alignment by ComSubStruct is compared with five structural alignment tools: CE [4], DALI [5], HMM-SA [2], FATCAT [6], and MATT [7].
 |
 |
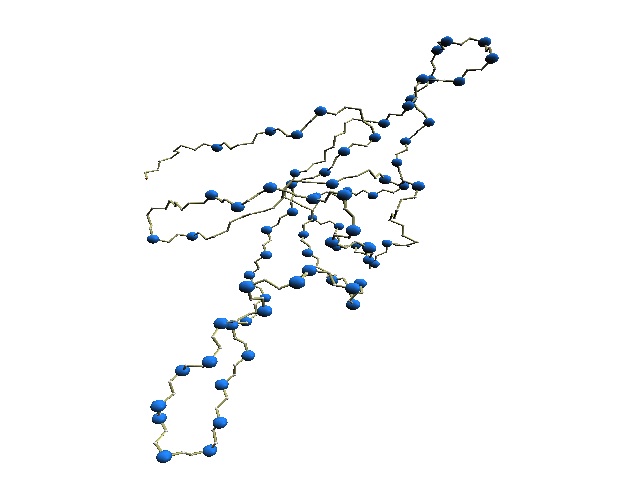 |
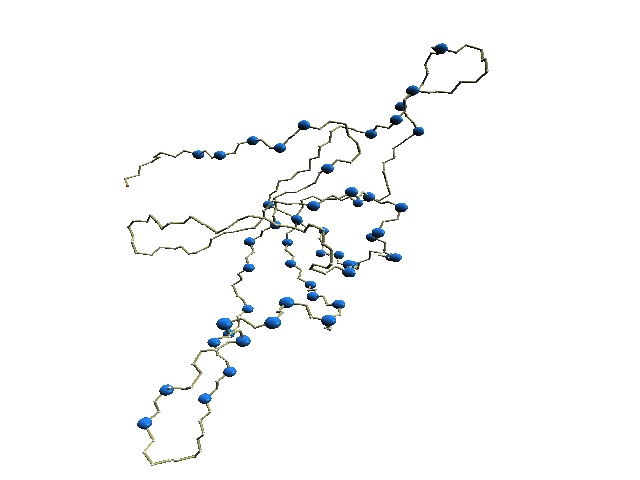 |
|||
| (A)
Structural variations of GroES. |
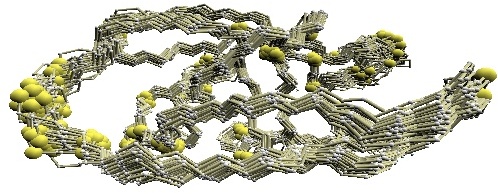
(B) D2 code-variable residues of GroES. |
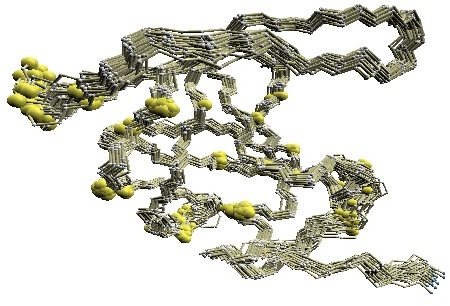
(C) HMM-SA-variable residues of GroES. | (D)
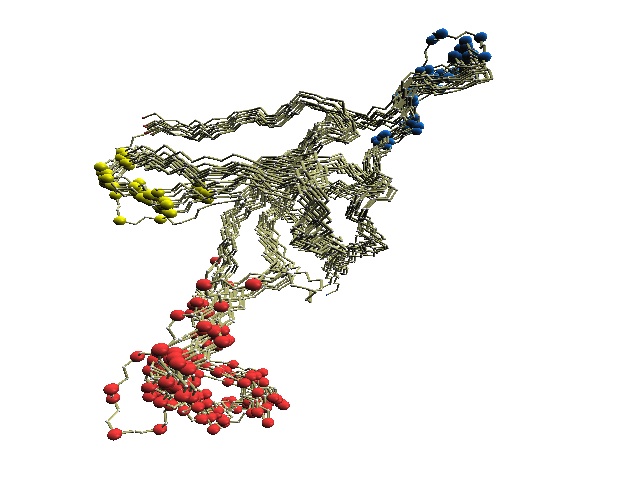
Stride-variable residues of GroES. |
(B) The 'master-slave' alignments by DALI with 1p3h-N as master, where residues with different D2-codes are colored accordingly: '0' in blue, 'A' in red, 'B' and 'Q' in orange, 'G' in green, and 'R' in yellow.
(C) The structure of 1p3h-N only is shown, where residues with different HMM-SA codes are shown in blue.
(D) The structure of 1p3h-N only is shown, where residues with different Stride codes are shown in blue.
References
[1] Pirovano W, Feenstra KA, Heringa J., The meaning of alignment: lessons from structural diversity. BMC Bioinformatics. 2008 Dec 23;9:556.
[2] Camproux AC, Gautier R, Tuffery P., A hidden markov model derived structural alphabet for proteins. J Mol Biol. 2004 Jun 4;339(3):591-605.
[3] Heinig, M., Frishman, D. (2004). STRIDE: a Web server for secondary structure assignment from known atomic coordinates of proteins. Nucl. Acids Res. , 32, W500-2.
[4] Shindyalov IN, Bourne PE. Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Eng. 1998;11:739–747.
[5] Holm L, Park J. DaliLite workbench for protein structure comparison. Bioinformatics. 2000;16:566–567.
[6] Ye Y, Godzik A. Flexible structure alignment by chaining aligned fragment pairs allowing twists. Bioinformatics. 2003;19:ii246–255.
[7] Menke M, Berger B, Cowen L. Matt: local flexibility aids protein multiple structure alignment. PLoS Comput Biol. 2008;4:e10.
[8] Roberts MM, Coker AR, Fossati G, Mascagni P, Coates ARM, Wood SP, Mycobacterium tuberculosis chaperonin 10 heptamers self-associate through their biologically active loops. J.BACTERIOL. 2003 185: 4172-4185.